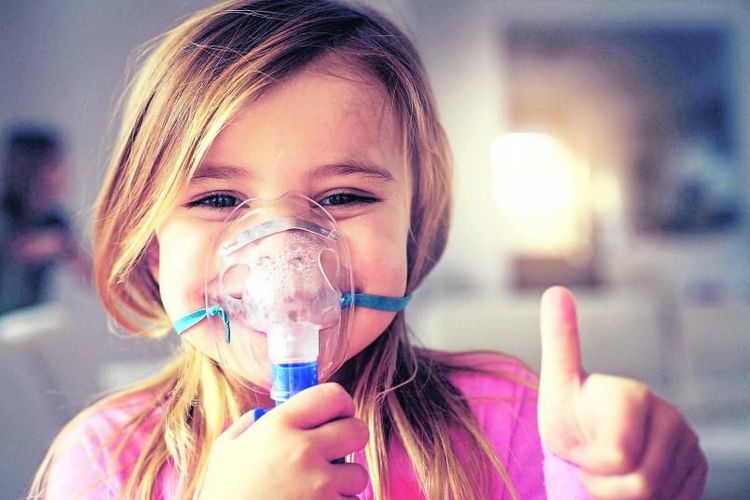
So selbstverständlich wie Zähneputzen ist für Kinder mit zystischer Fibrose das tägliche Inhalieren. Es gibt keine Ausnahmen.
Simon muss in die Kinderklinik. Man wird seinen Schweiß sammeln und den Kochsalzgehalt bestimmen. Ist die Konzentration zu hoch, lautet die Diagnose mit an Sicherheit grenzender Wahrscheinlichkeit zystische Fibrose. Dann würde ein defektes Gen in den Schleimhautzellen seines Körpers ein zähes Sekret bilden, das vor allem die Funktion von Lunge, Bauchspeicheldrüse und Verdauungstrakt beeinträchtigt. Dann würde unser Sohn für den Rest seines Lebens eine zeitintensive Therapie benötigen. Inhalieren müsste zu seiner täglichen Routine werden. So meine Befürchtung vor mittlerweile vier Jahren.
Simon macht sich zu dem Zeitpunkt keine Sorgen, er ist vier Jahre alt, chronische Erkrankungen sind nicht sein Thema. Er würde lieber im Park den Ball treten, lässt den Test im Spital aber geduldig über sich ergehen. Nach einer Stunde wird ihm das Behältnis mit dem Schweiß abgenommen. "Wenn der Test positiv ist, werden wir Sie morgen anrufen", sagt die Ärztin. Aber sie hätte nichts sagen müssen. Ich kannte das Prozedere, ich hatte es schon einmal erlebt. Bei Simons kleiner Schwester war die Erkrankung kürzlich diagnostiziert worden.
Drei Wochen zuvor hatte das Telefon geläutet. Wir warteten auf die Laborergebnisse. Ich stand unter der Dusche und hoffte auf einen gewöhnlichen Morgen. Frühstücken, Kinder anziehen. Zur Arbeit gehen. Um nicht unnötig in Angst versetzt zu werden, bat ich Familie und Freunde, während der Wartezeit auf Sarahs Resultate nicht anzurufen. Sobald die Ergebnisse da wären, würde ich mich melden, Entwarnung geben. Falscher Alarm, meine Tochter ist gesund, das wollte ich erzählen. Aber das Telefon läutete, vibrierte unablässig. Und hätte meine Frau den Anruf nicht entgegengenommen, vielleicht wäre dieses Telefon an der Wand zerschellt. Aber es hätte nichts geändert. Rund 25 Kinder werden jährlich in Österreich mit der Stoffwechselerkrankung zystische Fibrose, auch Mukoviszidose genannt, geboren. Unsere Tochter Sarah ist eines davon. Ihr älterer Bruder muss sich nun auch testen lassen, zur Sicherheit.
Am Anfang ein Fersenstich
Denn zystische Fibrose ist eine Erberkrankung. Da unsere Tochter betroffen ist, sind meine Frau und ich zweifellos, so wie jeder 25. Mensch in Europa, gesunde Mutationsträger. Statistisch betrachtet wird jedes vierte unserer Kinder erkrankt geboren. Wir hatten keine Ahnung. Nicht von unseren Erbanlagen, nicht von zystischer Fibrose. Die Dinge änderten sich, als meine Frau ein paar Wochen nach Sarahs Geburt einen Brief öffnete. Das durch einen Fersenstich erhobene Neugeborenen-Screening hatte eine Auffälligkeit ergeben, eine weitere Blutprobe sei erforderlich. Sorglos rief ich nach diesem neuerlichen Test im Labor an, bestimmt würde sich alles in Wohlgefallen auflösen – doch das Ergebnis war positiv. Sarah wurde zu einem Schweißtest gebeten. Ich wurde unwirsch, wollte Klarheit, nein, ich wollte beruhigende Worte. Aber ich bekam sie nicht zu hören.
An dieser Stelle begann sich das Puzzle zusammenzufügen. Die bescheidene Gewichtszunahme, die marmorierte Haut, der ständige Hunger, die schlaflosen Nächte – es passte. Ich wollte es nicht wahrhaben, suchte nach alternativen Erklärungen, aber im Grund begann ich an jenem Tag zu verstehen. Sarahs positiver Schweißtest war keine Überraschung, die Gewissheit traf unsere Familie trotzdem mit Wucht. Es waren außer Kontrolle geratene Emotionen. In den folgenden Tagen ging ich ohne Ambition in die Arbeit, Dienst nach Vorschrift, wenn überhaupt. Wie hätte ich noch eine Zeile schreiben können? Alles belanglos.
Das Wiener Allgemeine Krankenhaus stellte uns eine Psychologin zur Seite. Wir trafen uns in der Kinderklinik, ich stand am Fenster einer der obersten Etagen und starrte durch den Wiener Hochnebel. Dann drehte ich mich um, und als gäbe es nur einen einzigen Satz, den man hätte aussprechen können, eröffnete ich das Gespräch: "In meinem Kopf ist es schwarz."
Ich habe Fragen hinter mich gebracht. Allen voran die törichte Frage nach dem Warum. Unsere Tochter wird mit zystischer Fibrose leben, es ist nicht zu ändern, nur zu akzeptieren. Im Wiener AKH wies uns die spezialisierte Ambulanz den Weg aus der emotionalen Abwärtsspirale. Wir sprachen über Medikamente, Therapien und Ernährung. Wir sprachen über Ängste und Hoffnungen. Schrittweise wurden uns Informationen verabreicht, alle Fakten und Einschulungen wären auf einen Schlag kaum zu verarbeiten. Nach dem ersten Aufklärungsgespräch gab uns Sabine Renner, die Leiterin der Ambulanz, noch einen guten Tipp: "Lesen Sie nicht ziellos im Internet, das würde Sie beunruhigen. Viele Darstellungen sind nicht mehr aktuell." Ich ging nach Hause – und las ziellos im Internet.
Unsere Tochter blühte dank der vielen Kapseln, die sie nun täglich schlucken musste, auf. Man merkte ihr die Krankheit nicht mehr an. "Hauptsache gesund!", sagten Nichtsahnende. Erstaunlich, wie oft dieser Satz fällt. Was soll man darauf antworten?
Nach und nach klärten wir unser Umfeld auf: Familie, Freunde, Kollegen. Die Fragen wiederholten sich. Ist das heilbar? Nein. Wird das im Alter besser? Nein. Ist die Lebenserwartung eingeschränkt? Ja, schon. Der britische Cystic Fibrosis Trust geht davon aus, dass die Hälfte aller medizinisch versorgten Patienten über 41 Jahre alt wird. Was heißt das für Sarah? Es gibt nur eine vernünftige Antwort: nichts. Für individuelle Prognosen sind die Verlaufsformen zu unterschiedlich, selbst bei Geschwistern. Aber immerhin, die Zeiten, als zystische Fibrose ausschließlich Kinderärzte beschäftigte, sind vorbei, den sich stetig verbessernden Therapien sei Dank. Das Gesicht der Erkrankung hat sich verändert: Betroffene gehen auf die Universität, haben Jobs, sie gründen Familien. Der Australier Nathan Charles schaffte es 2014 sogar bis ins australische Rugby-Nationalteam.
Als würde man durch einen Strohhalm atmen: So wird das Gefühl der Kurzatmigkeit von den Patienten selbst häufig beschrieben. Dazu kommen Bronchitis, Lungenentzündung, Untergewicht. All diese Symptome sind fortwährend zu bekämpfen.
Einatmen, ausatmen
Die Inhalation nimmt bei der Behandlung einen hohen Stellenwert ein, sie wird zumindest zweimal täglich durchgeführt. Einerseits, um den zähen Schleim in den Atemwegen zu lösen, andererseits, um Medikamente direkt in die Lunge zu befördern. Die Einnahme verkapselter Enzyme soll die Unterfunktion der Bauchspeicheldrüse korrigieren und Mangelernährung vermeiden.
Zusätzlich nimmt Sarah Vitaminpräparate. Gezielte Antibiotikatherapie bekämpft bakterielle Infektionen. Wenn ich die Apotheke verlasse, bin ich bepackt wie ein Weihnachtsmann. Auch Sport und Bewegung sind wesentliche Faktoren zur Bewältigung der Erkrankung. In unserem Wohnzimmer steht ein Trampolin. Sarah hat mehr Sprünge in den Beinen als Olympiasieger Dong Dong (er heißt wirklich so!).
Um den intensiven Alltag mit den vielen neuen Prozeduren zu bewältigen, wurden wir in einer ersten Phase von einer mobilen Kinderkrankenschwester unterstützt. Lena war ein Glücksfall. Wenn ich in Selbstmitleid verfiel, rüttelte sie mich mit lebensnahen Weisheiten wach: "Es ist keine Katastrophe. Katastrophe ist, wenn Kind tot ist." Vielleicht könnte man es einen Hauch eleganter formulieren, aber wie soll man da widersprechen?

Noch ein Glücksfall: Sarah bockt nicht, sie macht mit, jeden Tag. Sie sagt Dinge wie: "Papa, wir müssen inhalieren!" Gut, das ist weniger ihrem Verantwortungsbewusstsein als der Vorfreude auf ein Fernsehprogramm, das sie währenddessen sehen darf, geschuldet, aber trotzdem: Sie macht mit, jeden Tag, stundenlang. Es gibt keine Ausnahme. Nicht im Urlaub, nicht zu Weihnachten, niemals. Sarah kennt es nicht anders.
Je früher zystische Fibrose behandelt wird, desto höher sind die Chancen auf eine hohe Lebensqualität. In diesem Sinne werden Kinder in Österreich seit 1997 über das Neugeborenen-Screening diagnostiziert. In Deutschland rang man sich erst im vergangenen Jahr zu diesem Schritt durch. Bis zur Behandlung ging oft wertvolle Zeit verloren.
Zystische Fibrose ist eine seltene Erkrankung. Weltweit wird die Anzahl der Patienten auf maximal 100.000 geschätzt. Die Inzidenz schwankt dabei von 1:1.800 in Irland bis zu 1:350.000 in Japan. In Österreich liegt sie bei rund 1:3.500. So oder so, zystische Fibrose ist kein Massenphänomen, dementsprechend klein das Interesse großer Unternehmen, in die Forschung zu investieren. Umso bemerkenswerter der Fortschritt: 2012 wurde in Europa das vom US-amerikanischen Hersteller Vertex Pharmaceuticals entworfene Medikament Kalydeco zugelassen. "Eine Wunderpille!", jubelten die Fachexperten.
Kleine blaue Tabletten
Das US-Nachrichtenmagazin "Forbes" sprach sogar von der "wichtigsten Arznei des Jahres". Der damalige US-Präsident Barack Obama wiederum würdigte den medizinischen Durchbruch in einer seiner Ansprachen zur Lage der Nation. In der Tat öffnete diese kleine blaue Tablette eine neue Perspektive. Erstmals wurde es möglich, zystische Fibrose kausal zu behandeln, zuvor zielte die Therapie auf Symptombekämpfung ab. Kalydeco lässt das Wasser strömen und hilft, den Schleim zu verflüssigen. Das bedeutet keine Heilung, die ursprünglichen Therapien müssen beibehalten werden, aber die Lungenfunktion verbessert sich, und die Patienten nehmen an Gewicht zu.
Der Haken an der Sache: Das Medikament wirkt in Mitteleuropa bei weniger als zwei Prozent aller Betroffenen, und zwar bei jenen, die eine Genmutation mit der Bezeichnung G155D aufweisen. Das 2015 zugelassene Nachfolgeprodukt Orkambi ist zwar weniger effektiv, kann aber bereits bei 50 Prozent aller Patienten eingesetzt werden. Ein Meilenstein in Sachen personalisierte Medizin. Er hat seinen Preis, die jährlichen Kosten belaufen sich auf 275.000 Euro pro Patient. Die Krankenkassen stöhnen, legen sich in manchen Ländern quer. "Don't put a price on our lives", antwortete eine irische Interessengemeinschaft. In Österreich wird Kalydeco ab dem zweiten Lebensjahr von der Krankenkasse übernommen, Orkambi ist ab dem zwölften Lebensjahr zugelassen.
Sarah hüpft derweil auf ihrem Trampolin, bläst Kugeln aus Styropor über den Tisch, lässt es im Wasserglas blubbern. Die Physiotherapie soll den Gasaustausch in der Lunge fördern. Sie soll helfen, den Schleim in den Atemwegen zu lockern. Wird das Sekret nicht abgebaut, bildet es einen Nährboden für Bakterien. Dann erkranken Patienten immer häufiger an Infektionen der Atemwege, die Lungenfunktion sinkt zusehends, eine Transplantation gilt als letzte Option. Für die wiederum ist Österreich kein schlechter Boden. Rund 120 Lungen werden pro Jahr im Wiener AKH verpflanzt, etwa zwanzig Eingriffe entfallen dabei auf Patienten mit zystischer Fibrose. Die durchschnittliche Wartezeit ist im internationalen Vergleich gering, der sogenannten Opt-out-Variante sei Dank. Gemäß der österreichischen Rechtslage dürfen Organe nach dem Tod entnommen werden, sofern man nicht zu Lebzeiten widersprochen hat. Und wer macht das schon? Um genau zu sein: 0,36 Prozent der Bevölkerung.
Ich saß also ein weiteres Mal vor dem Telefon, das drei Wochen zuvor geläutet hatte. Nun ging es um Simon. Zwar wurde er beim Screening für Neugeborene negativ getestet, aber ein Kind rutscht jährlich unter dem Radar durch. Unwahrscheinlich, ja, aber welche Bedeutung haben für uns noch Wahrscheinlichkeiten? Wird sich sein Leben grundlegend ändern? Ich küsse seine Stirn, sie schmeckt salzig, so wie man es von Kindern mit zystischer Fibrose kennt. Oder ist dieser Geschmack nur ein Produkt meiner Angst? Bilde ich mir das alles nur ein? Ich weiß es nicht mehr, ich sehe alles verschwommen. Gerade war alles im Lot, was passiert mit uns? Meine Frau nimmt das Telefon, sie will nicht mehr warten, sie ruft im Labor an. "Hat er es auch?" Die Antwort lässt kurz auf sich warten. "Nein, hat er nicht." (Philip Bauer, 25.2.2017)